전사체 : transcriptome
● 전사체 분석의 목적
- 표현형 질의 특성, 조절 기전의 이해
- 유전정보 기능 분석
- 질병 or 생물학적 기능 이상의 검출, 예측

● DNA가 아닌 RNA 발현을 연구하는 이유
- 기능적 연구, 유전자 발현량에 대해 알 수 있다.
- 일부 분자적 특성이 RNA에서만 나타난다. splicing, RNA editting
- 단백질 서열에 영향을 미치지 않는 돌연변이 해석 가능
- 단백질 코딩 체세포 돌연변이의 우선순위를 결정 가능
● 전사체 분석으로 알 수 있는 것
- 참조 유전체/전사체가 없을 때 새로운 전사체를 조립
- 새로운 isoform 검색
- 전사체 기능 검색과 분류
- 발현량 측정 및 서로 다른 환경에서 차등적으로 발현되는 유전자 분석
- 유전자의 온톨로지나 경로 확인
전사체 연구 발전 과정
sanger, microarray → NGS → no PCR NGS → single cell RNA-seq
NGS platform의 선택
- 종류마다 Data size, 길이 등 다르기 때문에 연구에 맞는 platform을 선택하는 것이 중요
NGS는 DNA, RNA, epigenetic, pritein 모두 사용 가능하다.
Microarray vs RNA-seq
1. Microarray
- cDNA를 이용하여 cell에 특정 RNA가 존재하는지를 확인/비교
- noncoding RNA도 가능

- 알려진 유전체 정보를 프로브로 디자인한다.
- 높은 background level로 발현량이 적은 유전체는 검출이 어렵다.
- 제한된 발현 범위(형광을 이용하기 때문에)
2. RNA-seq
- NGS 방법을 이용하여 발현 RNA를 검출한다.
- 낮은 발현의 유전자도 확인이 가능하다.
- 알지 못하는 유전자(새로운)의 발현도 측정 가능하다.
- 다양한 분석 연구에 활용 가능. Alternative Splicing, SNPs, gene fusion
- 최신 동향 : smRNAs, miRNA-seq, single-cell RNA-seq
RNA-seq 실험 디자인과 NGS
● RNA-seq pipe line
1. pre analysis
- 실험 디자인, seq-디자인, QC
2. core analysis
- transcriptome profiling, DEG, interprofiling
NGS -> preprocessing -> mapping -> estimate expression -> DEG selection -> Functional annotation
3. advanced analysis
- 시각화, 다른 seq과의 융합
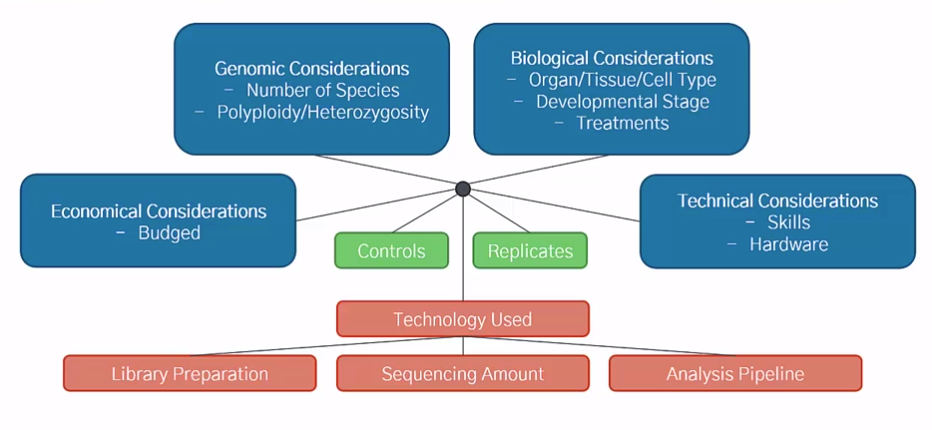
● 실험 디자인
고려 요인
1. 최종 목적
2. 어떤 성격의 실험군인가?
- control, repilcates 설정

3. 반복수
- 최소 3번, 많을수록 좋다.
- technical, biological 하게 오차가 생길 수 있기 때문이다.
4. cDNA library (RNA targeting)
5. 서열의 길이를 고려(isoform을 확인하기 위해 긴 것이 좋다.)
- SE, PE를 구분한다.
- SE : 단일가닥 seq, 잘 알려진 transcriptome이라면 short SE로 충분하다.
- PE : 양방향 seq, de novo transcript discover와 isoform 발현 분석에 유리하다.
6. sequencing depth
- 라이브러리를 얼마나 많이 읽을 것인가? (PCR 수준에서)
- sample이 많을수록 발경, 정량이 정확해지지만 off-target, noise 또한 증가
7. RNA 추출 및 quality check
● RNA-seq 분석의 유형
1. Referance genome-based
- 유전체 정보가 있는 경우
- transcript 재구성하는 능력이 크게 향상
- 대부분의 model organism
2. Referance genome-free
- de novo assemble 과정이 필요하다.
- RNA-seq 파라미터 의존적, 좋은 결과를 만들기 어렵다.
● Mapping의 3가지 전략
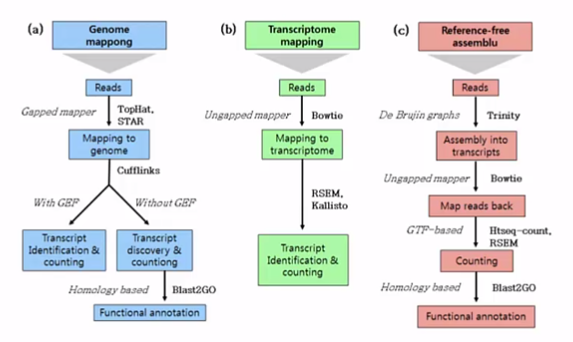
1. Referace genome-based
- 인트론 갭을 인정하는 알고리즘 사용(top hat, STAR)
-> prfiling(cufflinks) -> fuction annotation(Blast2 GO)
2. Referance-free but gene model
- 인트론에 대한 정보 x, 갭이 없어도 mapping 가능한 알고리즘 사용(Bowtio)
-> RSEM, Kallisto
3. Referance, gene model - free
- De novo assemble -> 다양한 알고리즘 존재(De Brujin graphs, Trinity) -> Bowtio -> RSEM
● 새로운 transcriptome 검출
- Short read를 이용한 novel transcript를 검출하는 것은 RNA-seq의 중요한 과제 중 하나
- Short read는 splice junction에 위치하는 경우가 적다.
- PE read와 높은 coverage는 소량 발현되는 유전자의 재구성에 유리하다.
- 반복 실험은 false-positive를 줄이기 위해 중요.
● read mapping result
- SAM/BAM 파일
- mapping 된 비율로 QC (refer: 85~95%, de novo: 70~%)
● 발현량 측정
- mapping 된 양으로 측정
- library가 크거나 exon이 많으면 더 크게 측정된다. -> gene 길이를 고려하는 수치를 이용
- RPKM/FPKM : 샘플 간, 유전자 간 발현량 비교
$$RPKM/FPKM = \frac{\text{total exon read}}{\text{mapping reads} \times \text{exon length}}$$
- TPM : RNA population에서 transcript length까지 고려
- TMM : 샘플 간 nomalization 방법 (단일 샘플 x)
▶ 차등발현 비교 전 실험 결과의 재현성을 확인해야 한다.
- 반복수에 따라 PCA그래프를 그려서 뭉쳐있는 지를 확인한다. cluster Dendrogram
DEG
● Fold - Change
$$FC = \frac{\text{expression in treatment}}{\text{expression control}}$$
- 주로 log를 취하여 사용한다.
- student's t-test, ANOVA, 2 way-ANOVA를 이용한다.
▶ 다중 비교 문제
- p <0.05에서 100번의 연구 중 5번의 오류가 발생한다. -> 최소화 하자
- 본페로니 FWER(보수적) -> multi-step 보정법(FDR) -> Volcano plot, MA plot을 비교한다.

▶ cluster 분석(HCL, k-means)
- 유전자 발현량을 기준으로 cell 군집화
● 차등 발현 유전자의 기능적 해석
- 다양한 DB를 이용한다.
▶ GSEA : 동일한 기능을 하는 Gene을 통계적으로 발굴
- 사전에 정의된 gene set
▶ GO : 유전자 정보가 든 DB, 세계적으로 공통
▶ DAVID : functional annotation을 위한 tool + infomatics
▶ Blast2GO : 사용 tool로 가장 많이 사용한다.(omicsBox), 기능 예측
● pathway 분석
▶ KEGG pathway : 네트워크와 관련된 DB
▶ Cytocape : 네트워크 시각화 소프트 웨어
▶ IPA : 많이 사용, 전문가 큐레이션 DB
'바이오 데이터 > 유전체 분석' 카테고리의 다른 글
| Microarray 데이터 전처리(RMA) (0) | 2021.03.17 |
|---|---|
| GEO 에서 데이터 다운받기(R) (0) | 2021.03.17 |
| GEO 에서 데이터 다운받기(web) (0) | 2021.03.17 |
| RNA-seq Technology (0) | 2021.03.12 |




댓글